42
logías como VIH/SIDA, diabetes, autoin-
munes, neurodegenerativas, virales o para
determinados tipos de medicamentos de-
signados como huérfanos.
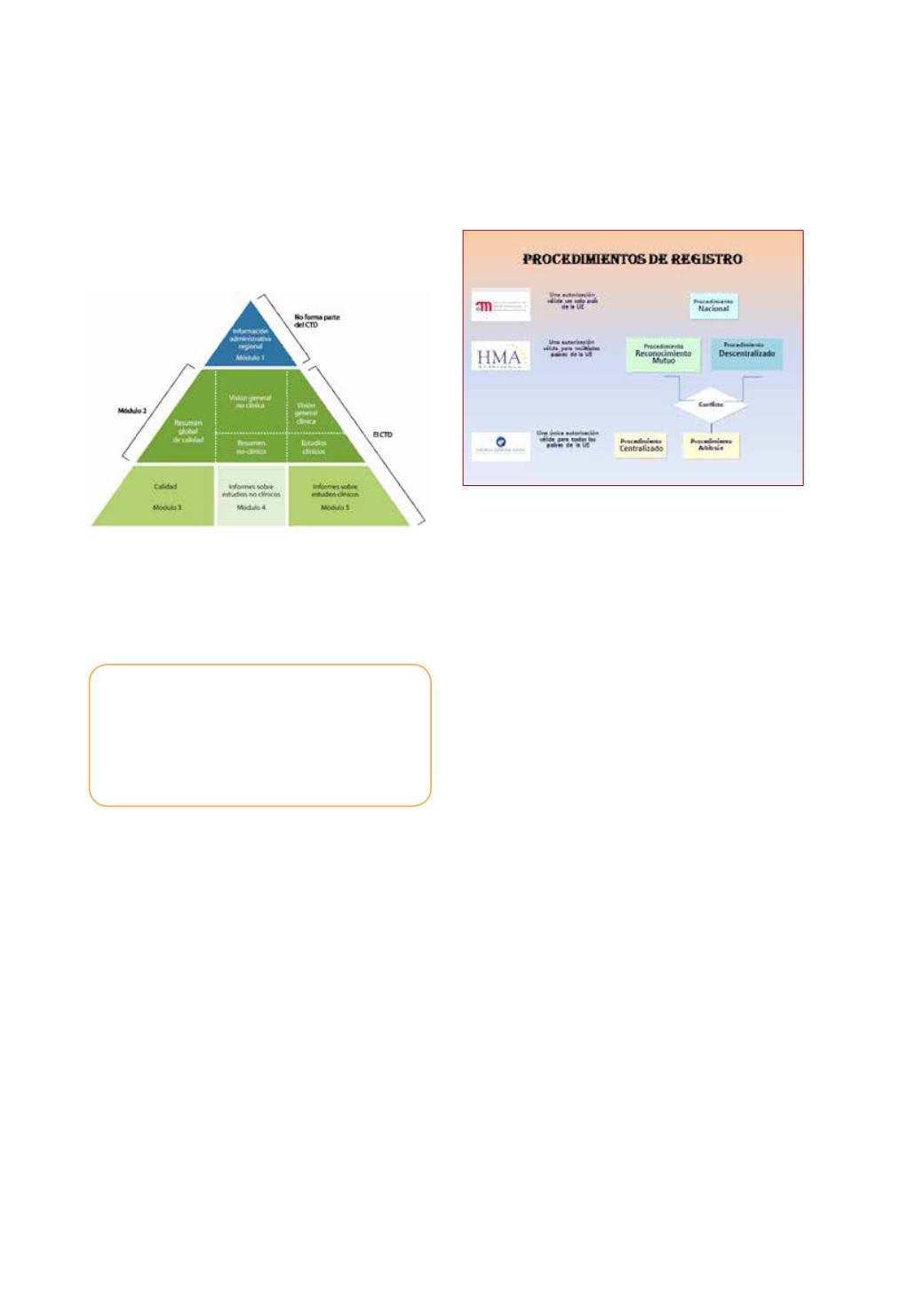
Otros procedimientos, responden a solici-
tudes de autorización para por ejemplo un
único país, al que se accede a través del de-
nominado “Procedimiento Nacional” o bien
mediante solicitudes para diversos países,
bien sobre la base de una autorización ini-
cial generada por un Estado miembro me-
diante el denominado “Procedimiento de
Reconocimiento Mutuo” o bien, por la soli-
citud de autorización para distintos países,
mediante el denominado “Procedimiento
Descentralizado” y que en ambos casos,
siempre se debe seleccionar un Estado
como Estado Miembro de Referencia.
Bases regulatorias comunitarias
sobre alergenos
Las Directivas 2001/83/CE
y su ulterior
modificación
2004/27/CE
del Parlamento
Europeo y del Consejo por la que se esta-
blece un código comunitario sobre los me-
dicamentos de uso humano, es clave para
establecer las bases regulatorias de los
alérgenos. En su artículo 1, apartado 4b,
incluye los alérgenos como medicamentos
inmunológicos y en el apartado 4b del mis-
mo artículo define los productos alergéni-
cos como cualquier medicamento desti-
nado a detectar o provocar una alteración
adquirida y específica de la respuesta in-
munológica a un agente alergizante y en su
artículo 2 de Ámbito de Aplicación, se eng-
De éste modo se va conformando el deno-
minado CTD, que en esquema organizado,
contiene la información y tratamiento de
los datos generados en las diferentes fases
del desarrollo del medicamento y que se
presentan a la agencia sanitaria para solici-
tar la autorización de comercialización.
Una vez que el medicamento ha sido auto-
rizado y se inicia su comercialización, se
accede a la denominada Fase IV, que con-
forma en términos generales las siguien-
tes características y funciones:
Marco regulatorio en la union eu-
ropea
El marco regulatorio en la Unión Europea,
dispone de varias modalidades o procedi-
mientos para obtener la autorización de co-
mercialización, bien de una forma automá-
tica para todos los países miembros o bien
mediante autorizaciones parciales o nacio-
nales. (Fig 1)
El procedimiento centralizado, coordinado
por la EMA, para su registro en todos los
Estados miembros, es obligatorio para de-
terminados tipos de medicamentos como
los enclavados como biotecnológicos (p.e.
ingeniería genética), terapias avanzadas
(p.e. terapia génica o celular) y por pato-
Fase IV
-El medicamento está comercializado.
-Se mantiene la observación sobre su
efectividad, seguridad y tolerancia en
condiciones de uso normales.
Regulación sanitaria de los alérgenos


